Integration im 1H-NMR Spektrum
Die NMR ist unter den gängigen spektroskopischen Methoden insofern einzigartig, als die Signalintensität direkt proportional zur Anzahl der Kerne ist, die das Signal verursachen (sofern bestimmte Bedingungen erfüllt sind). Mit anderen Worten: Alle Absorptionskoeffizienten für einen bestimmten Kern sind identisch. Aus diesem Grund werden Protonen-NMR-Spektren routinemäßig integriert, IR- und UV-Spektren dagegen nicht.
Ein typisches integriertes Spektrum von Ethylphenylsulfon ist unten abgebildet, zusammen mit einer NMR-spektroskopischen Analyse.:
1H-NMR (270 MHz, CDCl3) in ppm = 7.90 (d, 3J = X.X Hz, 2H, H4,6), 7.78 (m, 1H, H2), 7.66 (m, 3J = X.X Hz, 2H, H1,3), 3.51 (q, 3J = X.X Hz, 2H, H7), 1.19 (t, 3J = X.X Hz, 3H, H8). Anmerkung: die Kopplungskontanten werden in diesem Beispiel nicht behandelt.
Die Anzahl der verschiedenen Protonen wurde ausgehend von den Integrationssignalen mithilfe eines Lineals ermittelt.
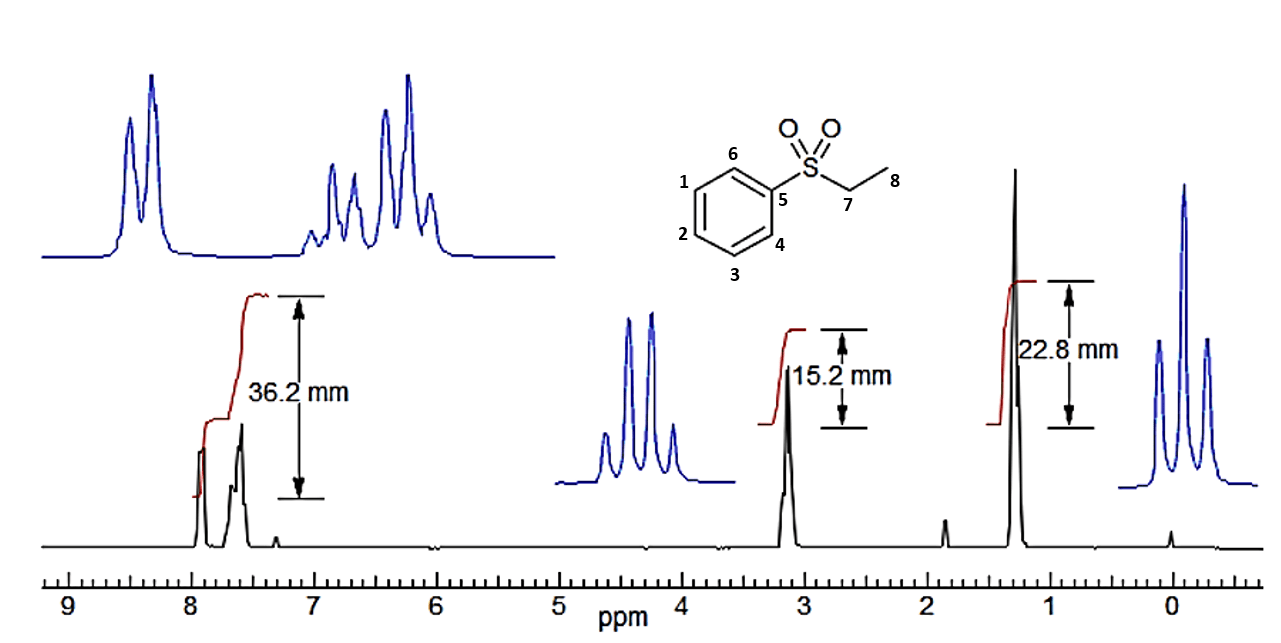 Abb.: 1H-NMR Spektrum - Ethylphenylsulfon
Abb.: 1H-NMR Spektrum - EthylphenylsulfonDie vertikale Verschiebung des Integrals gibt die relative Anzahl der Protonen an. Es ist nicht möglich, die absoluten Zahlen ohne zusätzliche Informationen (wie eine Summenformel) zu bestimmen. Wenn man im obigen Beispiel alle Integrale addiert, erhält man 74,3 mm. Dividiert man jedes Integral durch das kleinste (15,2 mm), erhält man ein Verhältnis von 2,38 : 1,0 : 1,50 für die drei Signale.
Die Multiplikation mit dem Nomierungsfaktor "zwei" ergibt ein Verhältnis von 4,76 : 2,0 : 3,03. Diese Integrationszahlen liegen nahe an denen für eine reine Verbindung mit dem erwarteten Verhältnis von 5 : 2 : 3. Es gibt jedoch nichts im Spektrum, was 10/4/6 oder höhere Vielfache ausschließt. Wenn wir eine Molekülformel haben (in diesem Fall C8H10O2S), ergibt die Division durch die Anzahl der Wasserstoffe 7,4 mm pro Wasserstoffatom.
Anschließend kann die Anzahl der Protonen für jedes Multiplett durch Aufrunden auf die nächste ganze Zahl bestimmt werden. Im Allgemeinen ist es möglich, Signale mit Intensitäten von 1 bis 10 oder so zuverlässig zu unterscheiden, aber es wird zunehmend schwieriger, eine korrekte Zuordnung vorzunehmen, wenn die Anzahl der Protonen in einem Multiplett über 10 hinausgeht, da die Methode inhärent ungenau ist.
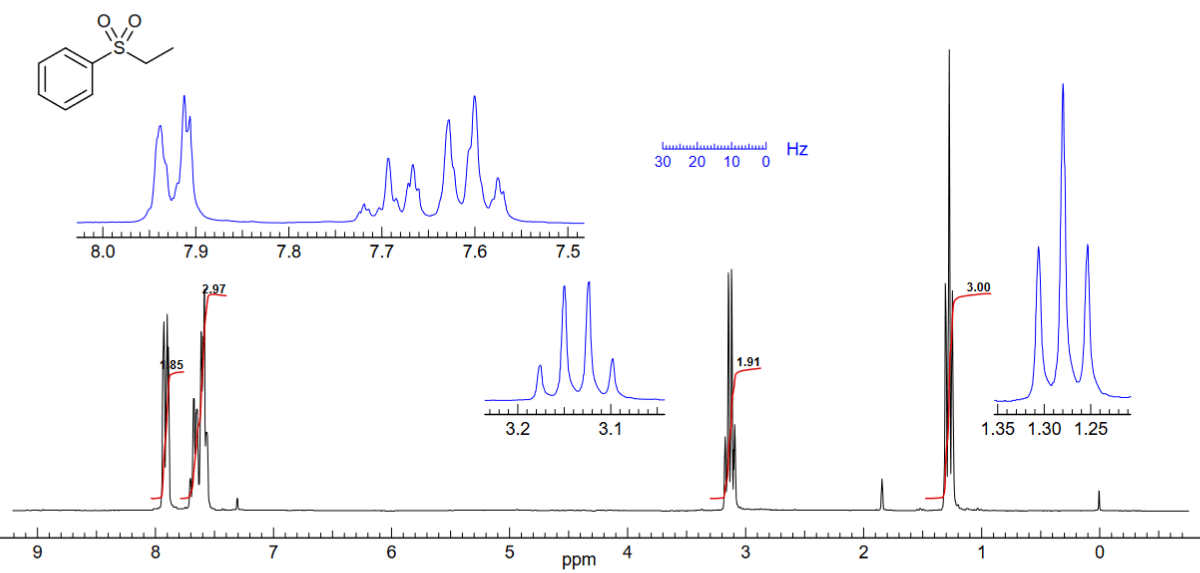
Die beiden Teile des aromatischen Protonenintegrals bei δ 7.6 und 7.9 können separat als ein 2 : 3-Verhältnis von ortho- zu meta+para-Protonen gemessen werden.
Wenn die Summenformel des Moleküls mit C8H10O2S gegeben ist, wissen wir, dass es 10 H-Atome in Molekülen haben.
Gesamtfläche: 36,2 + 15,2 + 22,8 = 74,2 mm, d.h. 7,4 mm pro H-Atom.
36,2 / 7,4 = 4,89 d.h. 5 H
15,2 / 7,4 = 2,05 d.h. 2 H
22,8 / 7,4 = 3,08 d.h. 3 H
Genauigkeit von Protonen-Integrationen
Die Integration von 1H-NMR-Spektren kann mit hoher Genauigkeit durchgeführt werden, doch ist dies nur möglich, wenn eine Reihe von Fehlerquellen ausgeschlossen werden. Mit modernen Spektrometern kann eine Genauigkeit von ±5 % leicht erreicht werden, wenn die Relaxationsprobleme richtig behandelt werden.
Um Fehler von <1 % zu erreichen, muss eine Reihe von Faktoren berücksichtigt und optimiert werden.
1. Signal-Rausch-Verhältnis. Das Spektrum muss ein angemessenes Signal-Rausch-Verhältnis aufweisen, um den für das Experiment erforderlichen Genauigkeitsgrad zu erreichen.
2. Sättigungseffekte. Die NMR-Spektroskopie zeichnet sich dadurch aus, dass die Relaxationsprozesse der Kernspins relativ langsam ablaufen (0.1 - 1 Sekunden ), verglichen mit Milli-, Mikro- und Pikosekunden bei IR- und UV-Methoden. Mit anderen Worten: Sobald das Spektrometer die Gleichgewichte der Kerne durch Scannen über die Resonanzfrequenz oder durch Pulsen der Kerne gestört hat, dauert es zwischen 0,1 und 100 Sekunden (typischerweise mehrere Sekunden), bis sie zu ihrer ursprünglichen Zustand zurückkehren (T1 die Spin-Gitter-Relaxationszeit). Wenn die Leistungseinstellungen zu hoch sind (bei CW-Spektren) oder der Pulswinkel und die Wiederholraten zu hoch sind (bei FT-Spektren), können die Spektren gesättigt und die Integrationen ungenauer werden, da die Relaxationsraten der verschiedenen Protonen in der Probe unterschiedlich sind. Sättigungseffekte sind besonders schwerwiegend bei kleinen Molekülen in mobilen Lösungsmitteln, da diese in der Regel die längsten T1-Relaxationszeiten haben.
Um zuverlässige Integrationen zu erhalten, muss das NMR-Spektrum so aufgenommen werden, dass eine Sättigung vermieden wird. Es ist nicht möglich, durch einfache Inspektion festzustellen, ob ein Spektrum in geeigneter Weise aufgenommen wurde. Es ist Sache des Anwenders, geeignete Vorkehrungen zu treffen (z. B. eine Pulsverzögerung von 5-10 Sekunden zwischen den Scans), wenn optimale Integrationen erforderlich sind. Glücklicherweise liefert auch ein Protonenspektrum, das ohne Pulsverzögerung aufgenommen wurde, in der Regel recht gute Integrationen (etwa innerhalb von 10 %). Es ist wichtig zu wissen, dass Integrationsfehler, die durch Sättigungseffekte verursacht werden, von den relativen Relaxationsraten der verschiedenen Protonen in einem Molekül abhängen. Die Fehler sind größer, wenn verschiedene Arten von Protonen verglichen werden (z. B. aromatischer CH mit einer Methylgruppe), als wenn die Protonen ähnlich oder identisch sind (z. B. zwei Methylgruppen).
3. Line-Shape Analyse. NMR-Signale in einem ideal abgestimmten Gerät haben die Form einer Lorenzlinie, d. h. die Intensität erstreckt sich über eine gewisse Strecke zu beiden Seiten des Peakzentrums. Die Integrationen müssen über einen ausreichend breiten Frequenzbereich durchgeführt werden, um genügend des Peaks für den gewünschten Genauigkeitsgrad zu erfassen. Beträgt die Peakbreite in halber Höhe 1 Hz, so ist eine Integration von ±2,3 Hz vom Zentrum des Peaks erforderlich, um 90 % der Fläche zu erfassen, ±5,5 Hz für 95 %, ±11 Hz für 98 % und ±18 Hz für >99 % der Fläche. Dies bedeutet, dass eng beieinander liegende Peaks mit der üblichen Methode nicht genau integriert werden können, sondern möglicherweise Liniensimulationen mit einem Programm wie NUTS oder WINDNMR erforderlich sind, um relative Peakflächen genau zu messen.
4. Digitale Auflösung. Ein Maximalwert muss durch eine ausreichende Anzahl von Punkten definiert werden, wenn eine genaue Integration erreicht werden soll. Die dabei auftretenden Fehler sind erstaunlich gering und erreichen 1 %, wenn eine Linie mit einer Breite in halber Höhe von 1 Hz alle 0,5 Hz abgetastet wird.
5. Isotopensatelliten. Alle C-H-Signale haben 13C-Satelliten, die ±JC-H/2 vom Zentrum des Peaks entfernt sind (JC-H ist typischerweise 115-135 Hz, obwohl Zahlen über 250 Hz bekannt sind). Sie müssen berücksichtigt werden, wenn eine Integration mit einer Genauigkeit von mehr als 99 % angestrebt wird. Größere Fehler entstehen, wenn die Satelliten eines nahen, sehr intensiven Peaks unter das zu integrierende Signal fallen. Die einfachste Methode zur Korrektur dieses Problems ist die 13C-Entkopplung, bei der die Satelliten auf den zentralen Peak komprimiert werden. Eine Reihe anderer Elemente haben signifikante Anteile von Spin-½-Kernen in natürlicher Häufigkeit, und diese erzeugen ebenfalls Satelliten, die groß genug sind, um die Integrationen zu stören.
13C-Satelliten haben auch eine positive Seite: Sie können als interne Standards für die Quantifizierung sehr kleiner Mengen von Isomeren oder Verunreinigungen verwendet werden, da ihre Größe im Verhältnis zum zentralen Peak genau bekannt ist.
6. Spinning-Seitenbänder. Diese können bei ± der Rotationsgeschwindigkeit in Hz in Spektren auftreten, die mit schlecht abgestimmten Spektrometern und/oder mit Proben in minderwertigen Röhren durchgeführt wurden. Sie ziehen die Intensität vom zentralen Peak ab. SSBs sind bei modernen Spektrometern selten signifikant.
7. Neigung und Krümmung der Grundlinie. Unter bestimmten Bedingungen können Spektren erhebliche Verzerrungen der Basislinie aufweisen, die eine qualitativ hochwertige Integration beeinträchtigen können. Standard-NMR-Aufbereitungsprogramme verfügen über Routinen zur Anpassung der Basislinie.
8. Entkopplung. Bei der Entkopplung, wie sie routinemäßig bei 13C-NMR-Spektren und gelegentlich bei 1H-NMR-Spektren durchgeführt wird, werden die Peakintensitäten durch nukleare Overhauser-Effekte (NOE) verzerrt. Die Integration solcher Spektren ergibt möglicherweise keine genauen Verhältnisse der Peakflächen.
Peak-Intensitäten. Unter bestimmten Bedingungen können auch Peakhöhen eine recht genaue Methode zur Quantifizierung sein. Wenn z. B. mehrere Singuletts verglichen werden, die alle die gleiche Linienbreite haben, und die Spektren so gemessen wurden, dass genügend Datenpunkte vorhanden sind, um die Linienform jedes Singuletts zu definieren, dann können Peakhöhen nützlich und unter idealen Bedingungen genauer sein als Integrationen.
Bestimmung der absoluten Mengen durch NMR-Integration. Obwohl NMR-Spektren im Prinzip dem Beer'schen Gesetz folgen, ist es schwierig (wenn auch nicht unmöglich), die absoluten Intensitäten von NMR-Spektren effektiv für die Quantifizierung zu nutzen (wie es routinemäßig bei UV- und manchmal IR-Spektren geschieht). NMR-Integrationen sind immer relativ. Daher muss ein interner Standard verwendet werden, um die Reaktionsausbeuten durch NMR-Integration zu bestimmen. Ein häufig verwendeter interner Standard für Protonen-NMR-Spektren ist Pentachlorethan - es ist flüssig, nicht allzu flüchtig und erscheint in einem Bereich des NMR-Spektrums (δ = 6.11 ppm), in dem es nur wenige Signale gibt. Es wird dringend empfohlen, die Verwendung von flüchtigen Materialien wie Dichlormethan, Chloroform oder Benzol etc. zu vermeiden, da es sehr schwierig ist, Verdampfungsverluste während des Transfers des Standards zu vermeiden, was zu falschen (hohen) Konzentrationen des Substrats führt.